ISO 13485:2016 Medical devices — Quality management systems — Requirements for regulatory purposes
ISO 13485:2016 specifies requirements for a quality management system where an organization needs to demonstrate its ability to provide medical devices and related services that consistently meet customer and applicable regulatory requirements. Such organizations can be involved in one or more stages of the life-cycle, including design and development, production, storage and distribution, installation, or servicing of a medical device and design and development or provision of associated activities (e.g. technical support). ISO 13485:2016 can also be used by suppliers or external parties that provide product, including quality management system-related services to such organizations.
Requirements of ISO 13485:2016 are applicable to organizations regardless of their size and regardless of their type except where explicitly stated. Wherever requirements are specified as applying to medical devices, the requirements apply equally to associated services as supplied by the organization.
The processes required by ISO 13485:2016 that are applicable to the organization, but are not performed by the organization, are the responsibility of the organization and are accounted for in the organization’s quality management system by monitoring, maintaining, and controlling the processes.
If applicable regulatory requirements permit exclusions of design and development controls, this can be used as a justification for their exclusion from the quality management system. These regulatory requirements can provide alternative approaches that are to be addressed in the quality management system. It is the responsibility of the organization to ensure that claims of conformity to ISO 13485:2016 reflect any exclusion of design and development controls.
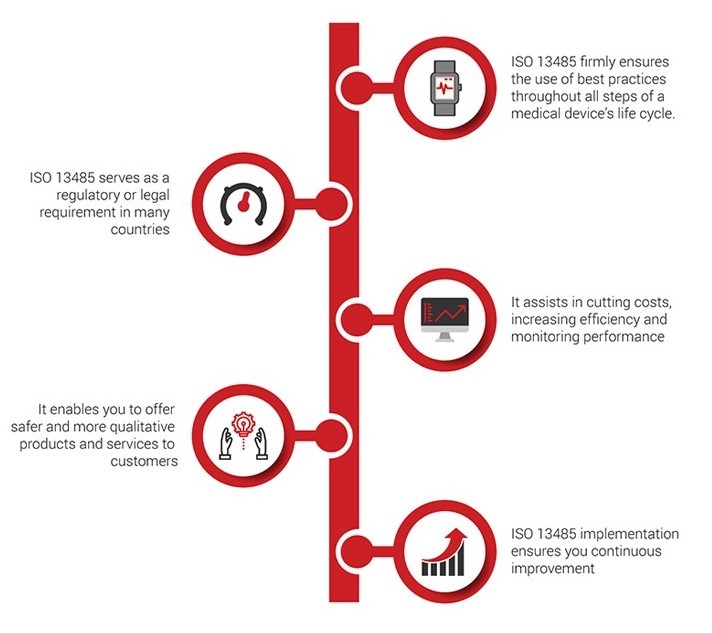
Benefits:
The benefits of ISO 13485 cannot be overstated. Companies of all sizes have realized significant savings in cost and time, plus other improvements that an efficient Quality Management System naturally bring about. The following is a list of six top reasons to implement ISO 13485 in your company:
Improve your company’s credibility and image. ISO 13485 is the internally recognized gold standard for quality in the medical device industry. Certification to this standard shows clients and customers that your company takes quality very seriously, and that you have a system in place to ensure it. Your QMS can be a powerful marketing tool, and it has become a requirement in many countries for suppliers to show compliance. This translates to more opportunity.
Improve customer satisfaction. The ISO 13485 standard is built on a set of quality management principles, one of which is ensuring customer satisfaction. This can be achieved by assessing customer needs and expectations, and striving to meet them. Customers know what they want, and what they need, and many of them will not even entertain a supplier that isn’t certified. Beyond that, ensuring the satisfaction of your existing customers keeps them coming back, and helps you sell your services to new customers. This translates directly to increased revenue.
Improve your processes. Using the process approach outlined in ISO 13485, it’s much easier to discover opportunities for improvements. You’ll be able to identify and eliminate waste within and between processes, reduce errors, and avoid rework—facilitating greater efficiency and cost savings.
Improve decision-making. Another quality management principle of ISO 13485 regards the use of evidence-based decision making. When you use facts and data to drive your decisions, those decisions tend to be better aligned with the strategic goals of your company. While “gut feelings” may be appropriate in some social situations, they can spell trouble in business. An added bonus is the increased insight into the health of your processes, and any improvements that are made, once you keep track of the data.
Create a culture of continual improvement. A third quality management principle making up the foundation of ISO 13485 is the concept of continual improvement. When adopted as the culture in your organization, management and staff will always be on the lookout for ways to improve on how things are done. By establishing systematic processes for reducing problems and mitigating their effects, everyone will spend less time cleaning up mistakes, and more time delivering quality products and services.
Better employee engagement. When employees are asked to help look for ways to improve their own processes, not only will they often provide the best insight – they will also be much happier and more invested in the success of the company. The more your employees understand their roles in delivering quality products and services, the more engaged they are, which leads to increased efficiency and productivity.
 ISO 13485:2016
ISO 13485:2016
Mandatory documents, procedures, and records:
Here are the documents’ list required for conformance with ISO 13485:2016:
- Roles played by the organization under relevant regulatory requirements – clause 4.1.1
- Procedure & records for the validation of the implementation of computer software – clause 4.1.6
- Quality Management System Manual – clause 4.2.2
- Records of Medical device file – clause 4.2.3
- Documented Procedure for document and data control – clause 4.2.4
- Documented Procedure for record control – clause 4.2.5
- Documented and communicated Quality Policy – clause 5.3
- Quality objectives (SMART) – clause 5.4.1
- Documented Roles, Responsibilities & authorities – clause 5.5.1
- Documented Procedure and records of management review – clause 5.6.1
- Documented Procedure and records of training – clause 6.2
- Documented Requirements for infrastructure and records of maintenance activities – clause 6.3
- Documented Requirements for the work environment – clause 6.4.1
- Documented requirements for control of contaminated or potentially contaminated product – clause 6.4.2
- Documented Process for risk management in product realization – clause 7.1
- Documented Outputs of product realization and records of product meeting realization requirements – clause 7.1
- Results of the customer requirements review and records of actions arising from it – clause 7.2.2
- Documented arrangements for communication with customers – clause 7.2.3
- Documented Procedure for design & development – clause 7.3.1
- Design & development planning documents – clause 7.3.2
- Design and development inputs records – clause 7.3.3
- Design and development outputs records – clause 7.3.4
- Design and development review records – clause 7.3.5
- Design verification plans, results, and conclusions records – clause 7.3.6
- Design validation plans, results, and conclusions records – clause 7.3.6
- Documented Procedure for transfer of design and development outputs to manufacturing – clause 7.3.8
- Documented Procedure and control of design and development changes records – clause 7.3.9
- Design and development file for each medical device type – clause 7.3.10
- Documented Procedure for purchasing – clause 7.4.1
- Establish Criteria and maintain evaluation and selection of supplier’s records – clause 7.4.1
- Verification record of procured product – clause 7.4.3
- Medical device each batch traceability records – clause 7.5.1
- Documented Requirements for cleanliness of product – clause 7.5.2
- Documented Requirements for medical device installation & establish acceptance criteria for verification of installation – clause 7.5.3
- Medical device installation records & Records of verification of installation – clause 7.5.3
- Documented Procedure for servicing of the medical device and its records – clause 7.5.4
- Sterilization process records – clause 7.5.5
- Documented Procedure of production and service provision process validation and its records – clause 7.5.6
- Documented Procedure of validation of a process for sterilization & sterile barriers systems and its records – clause 7.5.7
- Documented Procedure for product identification – clause 7.5.8
- Documented Procedure for traceability – clause 7.5.9.1
- Traceability, name, the address of the shipping package consignee records- clause 7.5.9.2
- Records on changes on customer property and its reporting to the customer – clause 7.5.10
- Documented Procedure for preserving the conformity of product – clause 7.5.11
- Documented Procedure for monitoring & measuring – clause 7.6
- Calibration records – clause 7.6
- Documented Procedure for validation of the application of computer software and its records – clause 7.6
- Documented Procedure for customer feedback and its records – clause 8.2.1
- Documented Procedure for complaint handling and its records – clause 8.2.2
- Regulatory authorities reporting records – clause 8.2.3
- Documented Procedure for internal audit – clause 8.2.4
- Audits records with its results – clause 8.2.4
- Record of the identity of the individual authorizing the release of the product – clause 8.2.6
- Documented Procedure of control of the nonconforming product and its records – clause 8.3.1
- Rework records – clause 8.3.4
- Documented Procedure of data analysis and its records – clause 8.4
- Documented Procedure of corrective action and its records – clause 8.5.2
- Documented Procedure of preventive action and its records – clause 8.5.3